Einleitung
Was ist AL-Amyloidose?
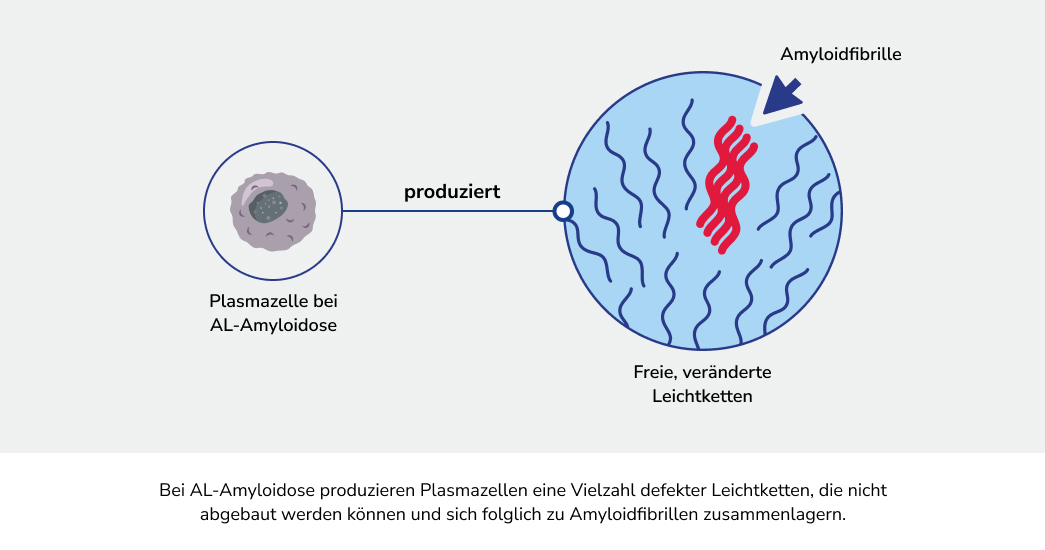
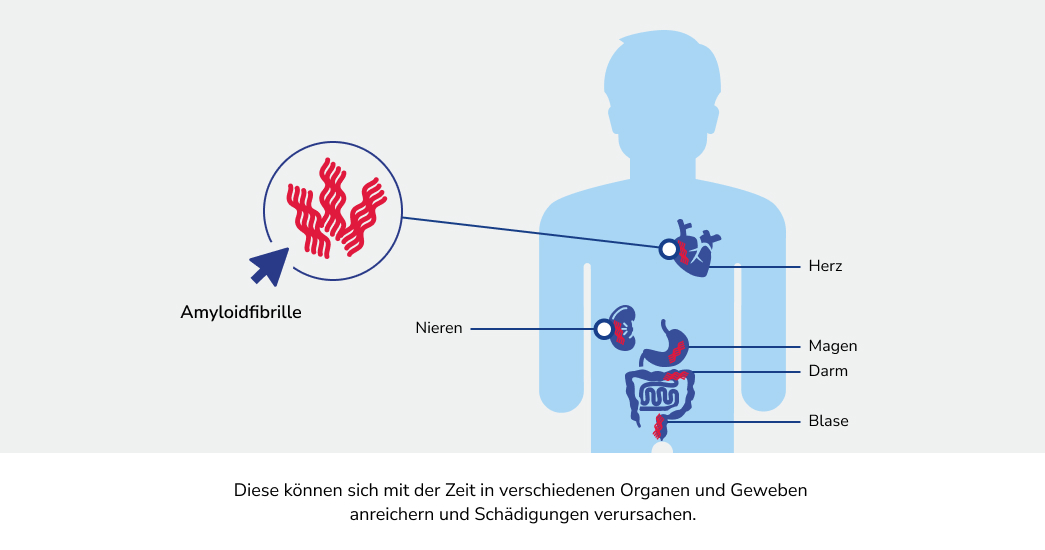
Die Amyloidose ist ein Überbegriff für verschiedene seltene Erkrankungen, bei denen sich bestimmte Eiweiße (Proteine) im Körper falsch falten. Die fehlgefalteten Proteine lagern sich als fadenförmige und unlösliche Strukturen, die sogenannten Amyloidfibrillen, im Körper ab.1
Es wird unterschieden zwischen lokalen Amyloidosen, bei denen nur ein Organ von den Ablagerungen betroffen ist und die örtlich beschränkt sind, und systemischen Amyloidosen, bei denen mehrere Organe betroffen sind.2 Die beiden häufigsten Formen der systemischen Amyloidose sind die Leichtketten-Amyloidose (AL-Amyloidose) und die Transthyretin-Amyloidose (ATTR-Amyloidose). Im Folgenden werden wir genauer auf die AL-Amyloidose eingehen.
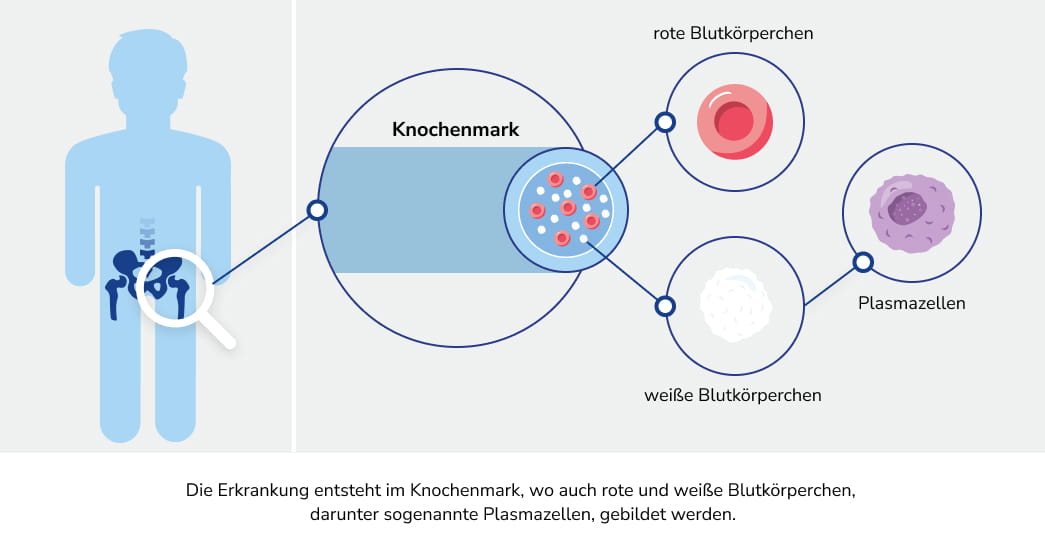
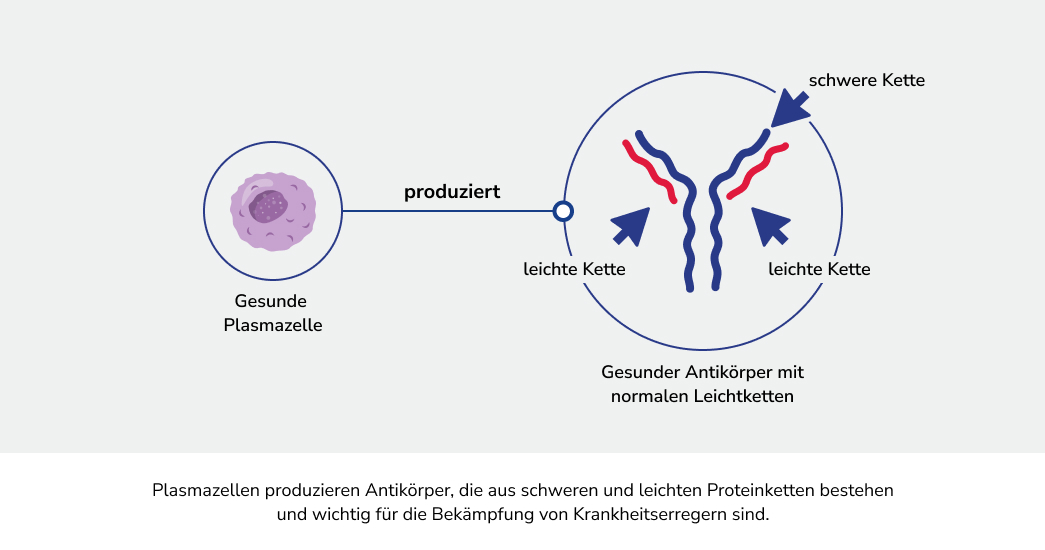
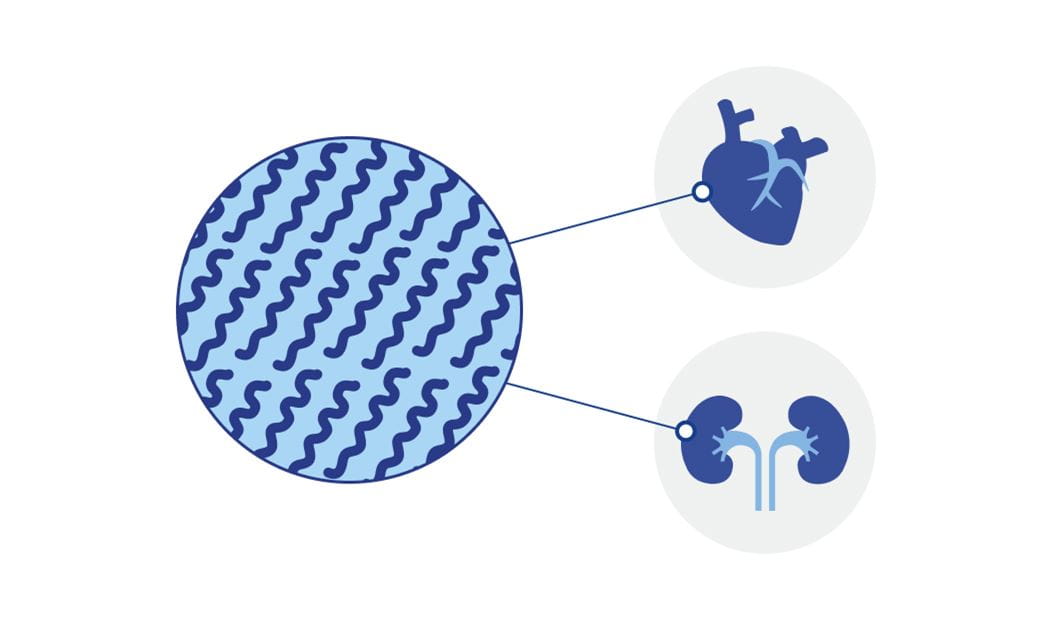
Bei der AL-Amyloidose vermehren sich bestimmte Zellen des Immunsystems, die einen Überschuss an fehlgefalteten Proteinen, den Leichtkettenproteinen, produzieren. Diese Proteine lagern sich als so genannte Amyloidfibrillen ab, welche sich fortlaufend in verschiedenen Geweben und Organen ansammeln und diese folglich schädigen können.1 Die resultierenden Symptome sind in der Regel zunächst unspezifisch. Dadurch ist die AL-Amyloidose nicht leicht festzustellen und es kann zur Verzögerung bei der Diagnose kommen.3,4 Es ist aber wichtig, die Erkrankung frühzeitig zu erkennen, um diese gut behandeln zu können.
Es wird unterschieden zwischen lokalen Amyloidosen, bei denen nur ein Organ von den Ablagerungen betroffen ist und die örtlich beschränkt sind, und systemischen Amyloidosen, bei denen mehrere Organe betroffen sind.2 Die beiden häufigsten Formen der systemischen Amyloidose sind die Leichtketten-Amyloidose (AL-Amyloidose) und die Transthyretin-Amyloidose (ATTR-Amyloidose). Im Folgenden werden wir genauer auf die AL-Amyloidose eingehen.
Bei der AL-Amyloidose vermehren sich bestimmte Zellen des Immunsystems, die einen Überschuss an fehlgefalteten Proteinen, den Leichtkettenproteinen, produzieren. Diese Proteine lagern sich als so genannte Amyloidfibrillen ab, welche sich fortlaufend in verschiedenen Geweben und Organen ansammeln und diese folglich schädigen können.1 Die resultierenden Symptome sind in der Regel zunächst unspezifisch. Dadurch ist die AL-Amyloidose nicht leicht festzustellen und es kann zur Verzögerung bei der Diagnose kommen.3,4 Es ist aber wichtig, die Erkrankung frühzeitig zu erkennen, um diese gut behandeln zu können.